
Prof. R. Sowdhamini - Genome analysis
Amino acid sequences of proteins emerging from genome sequencing projects are examined using several sequence analysis tools with an aim of recognising their structure and function. Hidden Markov models (Eddy, 1998; Hargbo & Eloffson, 1999), PSI-BLAST profiles (Park et al., 1998; Teichmann et al., 2000) are some techiques employed to obtain consensus prediction for proteins which are considered as 'hypothetical' or 'unknown'. One expects negligible sequence identity among superfamily members that are related in the twilight zone. Searches techniques are always not capable of establishing the distant relationship.
Signatures that could be characteristic to protein families, whose presence can guide a sequence to adopt that particular fold and function is an useful approach to establish distant homology among proteins. To identify motifs for superfamilies, search for homologues members have been undertaken and conserved regions have been determined. Conserved interacting motifs for different superfamilies are listed for individual members. Regions that are present at equivalent positions in structure among the superfamily members have been assigned as Interacting motifs of representing the superfamilies. Searches against the sequence databases have been performed. We establish the motif based sequence search approach is more sensitive and has higher coverage compared to other contemporary methods.(Bhaduri et al 2003). The motif based search is now being employed to search genome sequence databasesfor identifying remote homologues and analysis superfamily distribution.
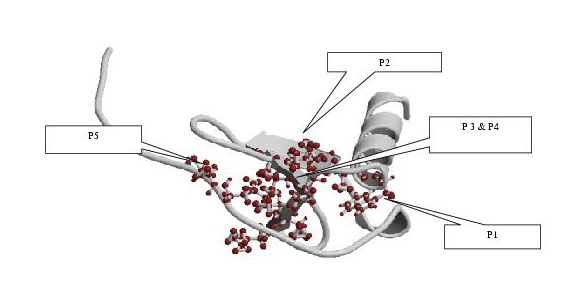
Figure1:Interacting motifs in interleukin-8-like (il8-like) superfamily
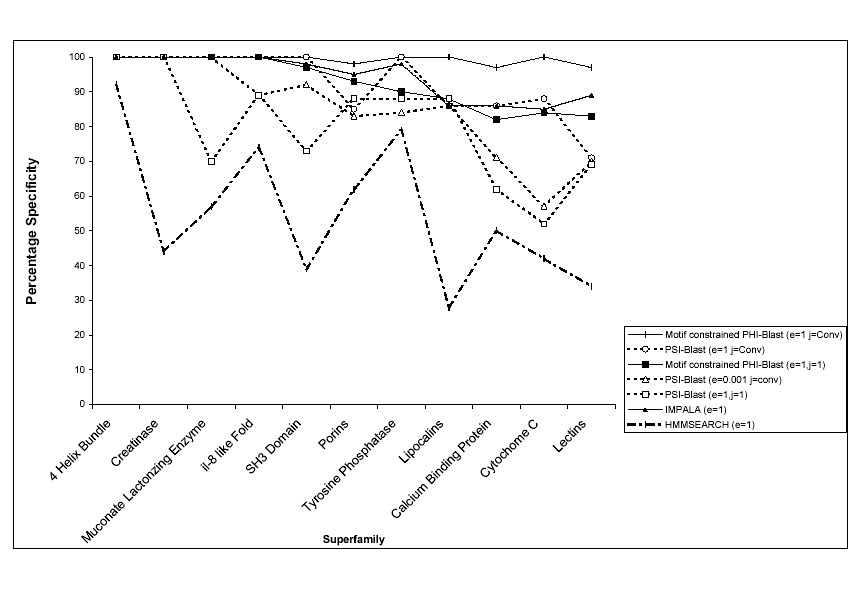
Figure2:Benchmarking motif-constrained approach by searching for 11 superfamilies against protein structural database. The percentage specificity is compared with other sensitive sequence search methods.
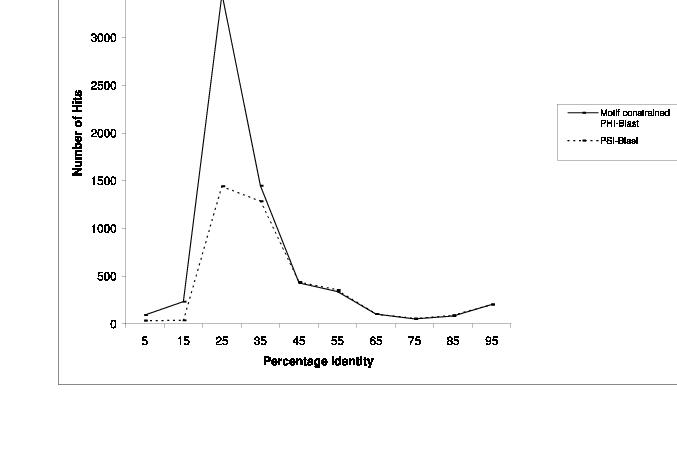
Figure3:The range of percentage identity where reliable connections of non-redundant sequence database to pre-existing families could be obtained using different methods.
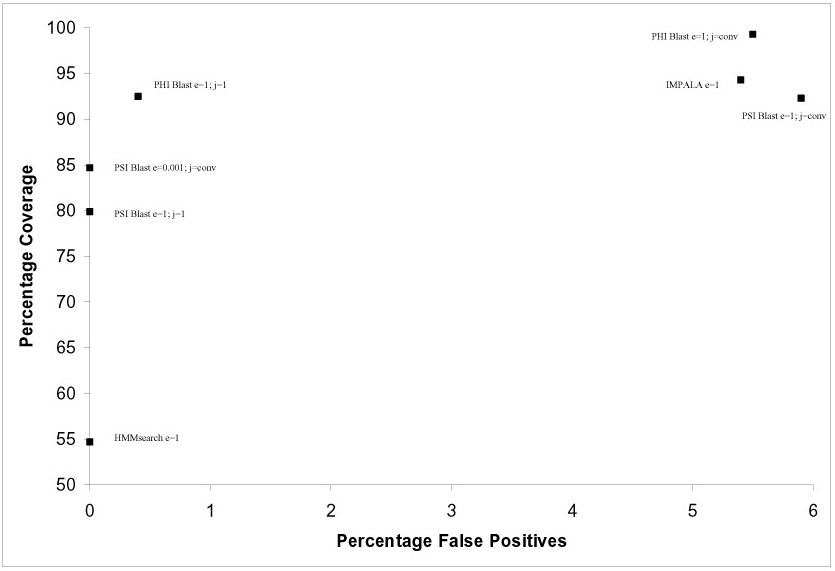
Figure4:Distribution of coverage and rate of false positives for 11 superfamilies against 93 genome databases using PSI-BLAST and motif-constrained approach.
Enhanced function annotations for Drosophila serine proteases: a case study for systematic annotation of multi-member gene families.
Systematically annotating function of enzymes that belong to large protein families encoded in a single eukaryotic genome is a very challenging task. We carried out such an exercise to annotate function for serine-protease family of the trypsin fold in Drosophila melanogaster, with an emphasis on annotating serine-protease homologues (SPHs) that may have lost their catalytic function. Our approach involves data mining and data integration to provide function annotations for 190 Drosophila gene products containing serine-protease-like domains, of which 35 are SPHs. This was accomplished by analysis of structure-function relationships, gene-expression profiles, large-scale protein-protein interaction data, literature mining and bioinformatic tools. We introduce functional residue clustering (FRC), a method that performs hierarchical clustering of sequences using properties of functionally important residues and utilizes correlation co-efficient as a quantitative similarity measure to transfer in vivo substrate specificities to proteases. We show that the efficiency of transfer of substrate-specificity information using this method is generally high. FRC was also applied on Drosophila proteases to assign putative competitive inhibitor relationships (CIRs). Microarray gene-expression data were utilized to uncover a large-scale and dual involvement of proteases in development and in immune response. We found specific recruitment of SPHs and proteases with CLIP domains in immune response, suggesting evolution of a new function for SPHs. We also suggest existence of separate downstream protease cascades for immune response against bacterial/fungal infections and parasite/parasitoid infections. We verify quality of our annotations using information from RNAi screens and other evidence types. Utilization of such multi-fold approaches results in 10-fold increase of function annotation for Drosophila serine proteases and demonstrates value in increasing annotations in multiple genomes.
Domain architectural census of eukaryotic gene products containing O-protein phosphatases.
Intricate molecular signalling within cellular environment is manifested through phosphorylation of proteins. Regulation of the phosphorylation state is executed through complex networking among kinases and their biochemical antagonists, the protein phosphatases. Protein dephosphorylation in eukaryotic systems is largely performed through four structurally distinct Ser/Thr and Tyr O-protein phosphatase superfamilies. 555 O-protein phosphatases, belonging to the four distinct families, could be identified using sensitive sequence search techniques across five eukaryotic model organisms (yeast, fly, worm, mouse and humans). These phosphatases could be grouped into 49 subfamilies associated with distinct domain architecture and discrete biochemical function. Only five of the architectures are shared across the five eukaryotic genomes. Interestingly, the number of occurrence of tyrosine phosphatases is correlated to the complexity of the genome. Analysis of domain architectures suggests amenability of the tyrosine phosphatases to occur in complex architectures unlike Ser/Thr phosphatases. Domain duplication and shuffling is shown as the customary mechanism for the evolution of the phosphatases. Several architectures are common between humans and other genomes, which are probably non-linearly inherited in humans or specifically lost in several others.
Cross genome phylogenetic analysis of human and Drosophila G protein-coupled receptors: application to functional annotation of orphan receptors.
BACKGROUND: The cell-membrane G-protein coupled receptors (GPCRs) are one of the largest known superfamilies and are the main focus of intense pharmaceutical research due to their key role in cell physiology and disease. A large number of putative GPCRs are 'orphans' with no identified natural ligands. The first step in understanding the function of orphan GPCRs is to identify their ligands. Phylogenetic clustering methods were used to elucidate the chemical nature of receptor ligands, which led to the identification of natural ligands for many orphan receptors. We have clustered human and Drosophila receptors with known ligands and orphans through cross genome phylogenetic analysis and hypothesized higher relationship of co-clustered members that would ease ligand identification, as related receptors share ligands with similar structure or class.
RESULTS: Cross-genome phylogenetic analyses were performed to identify eight major groups of GPCRs dividing them into 32 clusters of 371 human and 113 Drosophila proteins (excluding olfactory, taste and gustatory receptors) and reveal unexpected levels of evolutionary conservation across human and Drosophila GPCRs. We also observe that members of human chemokine receptors, involved in immune response, and most of nucleotide-lipid receptors (except opsins) do not have counterparts in Drosophila. Similarly, a group of Drosophila GPCRs (methuselah receptors), associated in aging, is not present in humans.
CONCLUSION: Our analysis suggests ligand class association to 52 unknown Drosophila receptors and 95 unknown human GPCRs. A higher level of phylogenetic organization was revealed in which clusters with common domain architecture or cellular localization or ligand structure or chemistry or a shared function are evident across human and Drosophila genomes. Such analyses will prove valuable for identifying the natural ligands of Drosophila and human orphan receptors that can lead to a better understanding of physiological and pathological roles of these receptors.