
Prof. Sumantra Chattarji - Earlier Work
Neural plasticity in the amygdala: implications for stress disorders
Some experiences are memorable, others forgettable. How does a particular experience leave its mark as a memory in the brain? For more than a century, the search for a biological basis of memory formation has centered on the synapse, the junction where information is passed from one brain cell to another. The remarkable ability of synapses to change in response to experience, a property described as “synaptic plasticity”, is believed to mediate long-term storage of information in the brain. Although common cellular and molecular mechanisms underlying synaptic plasticity and memory have been identified over the past few decades, we know little about why some memories last for a long time while others fade away. My laboratory is interested in understanding why memories of emotional events are often very powerful and persistent. Why do war veterans or victims of severe stress continue to have vivid flashbacks of traumatic events from their past, while their cognitive abilities diminish? Stress disorders bring these questions into sharp focus because chronic stress has contrasting effects on different types of memories. Stress impairs memories of facts and events, which depend on synaptic plasticity in a brain structure called the hippocampus. In contrast, stress greatly amplifies emotional memories, which are processed by another structure called the amygdala. But little is known about the neural basis for this contrast.
Therefore, we study the effects of stressful experiences on synapses, cells and microcircuits in the hippocampus and amygdala, by using a combination of behavioral, neuroanatomical, computational, genetic engineering and electrophysiological techniques. Using this strategy, we have identified several novel neural correlates of stress-induced plasticity in the amygdala, which are strikingly different from those observed in the hippocampus. Our findings suggest that prolonged stress leaves its mark by enhancing both the physiological and structural basis of synaptic connectivity in the amygdala, thereby triggering the emotional symptoms observed in stress-related psychiatric disorders.
Image
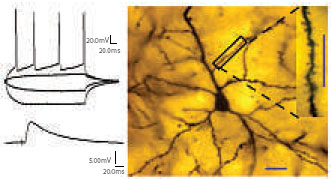 |
Left: Action potentials (top) and synaptic response (bottom) recorded from a pyramidal neuron in the lateral amygdala. Right: Dendrites and spines (inset) of a pyramidal cell in the lateral amygdala. |
1. Neural correlates of stress-induced plasticity in the amygdala
We are investigating the neural basis for the striking differences between hippocampal and amygdaloid function by characterizing the effects of stress on these two brain areas at multiple levels of neural organization spanning behavior, circuits, cells, synapses, and molecules.
a. Quantification of structural plasticity
Rupshi Mitra, Ajai Vyas, Shantanu Jhadav, Anupratap Tomar and Amrita Kuthiala
It has long been hypothesized that morphological and numerical alterations in dendritic spines underlie long-term structural encoding of experiences. We have investigated the efficacy of aversive experience, in the form of acute (AIS) and chronic immobilization stress (CIS), in modulating spine density in the basolateral amygdala (BLA) of male rats. We find that CIS elicits a robust increase in spine density across both primary and secondary branches of BLA spiny neurons (Figure 1 a & b). We observed this CIS-induced spinogenesis in the BLA one day after the termination of CIS. In contrast, AIS fails to affect spine density or dendritic arborization, when measured one day later. Strikingly, the same AIS causes a gradual increase in spine density ten days later, but without any effect on dendritic arbors. However, unlike CIS-induced spine formation, the gradual increase in spine density 10 days after a single exposure to AIS is localized closer to the cell soma (Figure 1 c). These findings demonstrate for the first time that the duration of stress can modulate the spatiotemporal pattern of spine formation in the BLA, which is believed to be a locus of storage for fear memories.
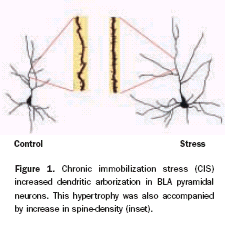 |
| Figure 1: Stress duration modulates the spatiotemporal pattern of spinogenesis in BLA. |
b. Animal behavioral studies
Ajai Vyas, Anup G. Pillai, Anupratap Tomar, Laxmi T. Rao and Aparna Suvrathan
The above findings, taken together with our earlier observations on stress-induced dendritic growth, suggest that stress leads to a significant strengthening in the overall synaptic connectivity of BLA neurons. This could have a profound facilitating impact on the network output of the amygdala in terms of emotional behavior. Hence we are investigating the behavioral consequences of stress-induced morphological changes by studying the impact of stress on various animal models of anxiety, depression, and fear. Exposure to 10 days of CIS facilitates anxiety-like behavior in the elevated plus-maze and this enhanced anxiety persists for up to 21 days after the termination of CIS. Our results also indicate that the long-lasting increase in CIS-induced anxiety is accompanied by a persistent increase in dendritic arborization of BLA spiny neurons. Strikingly, BLA hypertrophy is different from hippocampal CA3 atrophy, which is reversible within the same 21-day period of stress-free recovery. In another study we found that tianeptine, an antidepressant, prevents stress-induced increase in anxiety and BLA dendritic remodeling. Our results provide the first evidence for a pharmacological intervention that is effective in reversing the effects of chronic stress on structural plasticity of BLA dendrites as well as anxiety-like behavior.
More recently, we have been exploring potential links between stress-induced spinogenesis and emotional behavior. The delayed and localized formation of BLA spines (section a), after a single episode of acute stress (AIS), is paralleled by a gradual development of anxiety-like behavior on the plus-maze 10 days after AIS (Figure 1 d). Taken together these data suggest that stress may facilitate symptoms of chronic anxiety disorders by enhancing synaptic connectivity in the BLA. These studies are now being extended to explore the impact of fear learning on amygdalar spine plasticity.
Collaborators: Bruce McEwen, Rockefeller University, USA; Carmen Munoz, Servier, France.
c. In vitro electrophysiological recordings from single neurons
Sharath Bennur and Aparna Suvrathan
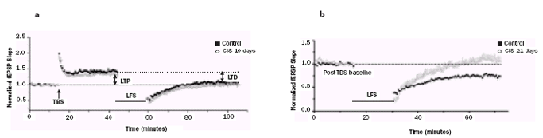
Over the years, research on the cognitive deficits caused by chronic stress has focused primarily on the hippocampus. These studies have established two cellular correlates of stress-induced plasticity in hippocampal pyramidal neurons – dendritic atrophy and impaired long-term potentiation (LTP), a candidate mechanism of synaptic plasticity mediating learning and memory. Importantly, the N-methyl-D-Aspartate (NMDA) subtype of glutamate receptors has been implicated in both forms of stress-induced hippocampal plasticity. Stressinduced impairment of LTP is believed to underlie the debilitating effects of severe stress on hippocampal learning and memory. Amygdala-dependent fear and anxiety, on the other hand, is facilitated by chronic stress. But little is known about the synaptic basis for this striking contrast. We find that chronic stress, in addition to reducing inhibition and eliciting intrinsic plasticity, triggers metaplasticity by enhancing the relative contribution of NMDA receptors to synaptic transmission in the lateral amygdala (LA) (Figure 2). This stress-induced increase in NMDA receptor function (Figure 2 e & f ), in turn, leads to enhanced LTP (Figure 2 c & d). These findings, apart from highlighting differences with respect to the hippocampus, suggest that prolonged stress primes the amygdala by facilitating subsequent induction of synaptic plasticity, thereby creating an ideal synaptic substrate for greater anxiety and fear.
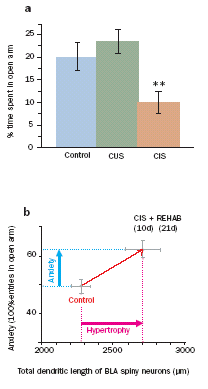 |
Figure 2: Chronic stress facilitates NMDA receptor function (e & f ) and LTP (c & d) in pyramidal neurons (b) in the LA (a). |
d. In vivo electrophysiological recordings in f reely behaving animals.
Laxmi Rao, Supriya Ghosh and Anupratap Tomar
In view of evidence for contrasting patterns of stress-induced plasticity in the amygdala and hippocampus, we are investigating the network correlates of stress-induced modulation in amygdalar and hippocampal output with in viv o recordings using chronically implanted electrodes in freely behaving rats. We find that theta activity (6-10 Hz) is enhanced one day after CIS in both the LA and hippocampus. A similar increase is also observed in LA 2 hours after AIS, but not a day later. Cross-correlation analyses reveal a gradual increase in correlated theta activity over a 21day a period of stress-free recovery after CIS. This enhanced synchrony is evident only between LA and CA3, two areas which exhibit the most robust stress-induced structural plasticity. Our results suggest that the duration of stress can have contrasting effects on the temporal dynamics of neural activity in the amygdala and hippocampus. The temporal sequence and spatial localization of these patterns of activity, in turn, may be an important determinant of stress-induced plastic changes in the hippocampus and amygdala.
e. Computational models
Rishikesh Narayanan and Anusha Narayan
Dendritic atrophy and impaired LTP have been identified as key measures of chronic stress-induced plasticity in the hippocampus. It has been hypothesized that these disparate structural and physiological correlates of stress lead to hippocampal dysfunction by reducing postsynaptic dendritic surface, thereby adversely affecting the availability of synaptic inputs and suppressing LTP. We performed computational simulations to examine the validity of this framework using a biophysical model of hippocampal CA3 pyramidal neurons (Figure 3 a). We find that stress-induced atrophy of CA3 dendrites amplifies intrinsic excitability at both the single-neuron and network levels. Moreover, coupled with experimentally observed modulation of NMDA receptor currents, such intrinsic plasticity tilts the balance of plasticity mechanisms in favor of synaptic potentiation over depression. Thus, paradoxically, our results suggest that stress may impair hippocampal learning and memory, not by directly inhibiting LTP, but because of the imbalance of bidirectional synaptic plasticity caused by stress-induced facilitation of intrinsic and
synaptic excitability.
 |
| Figure 3: Computational models of single neurons in the hippocampus (a) and amygdala (b). Stress causes hyperexcitability in atrophied CA3 pyramidal neurons (a). Model of different spiking properties exhibited by LA neurons (b). |
We are now extending these studies to build models of cells located in various subdivisions of the lateral amygdala that have been experimentally distinguished based on their morphological and physiological characteristics (Figure 3b). We find that variations in intrinsic firing properties among these neuronal classes may be controlled by active membrane properties rather than being a direct reflection of the morphological differences. We are also exploring the validity of the hypothesis that neurons in the ventral subregions of the lateral amygdala could be involved in integration a nd storage of higher order emotional information. Predictions from these computer models, in turn, can be tested using electrophysiological recordings described above.
Collaborator: Joseph LeDoux and Luke Johnson, New York University, USA
2. Molecular targets of stress disorders: studies using genetically engineered mice
a. The role of brain-derived neurotrophic factor (BDNF) in anxiety and depression.
Sumantra Chattarji
Structural changes in the hippocampus, mediated by reduced neurotrophin signaling, has been proposed as an underlying cause of depression. Using transgenic overexpression of the neurotrophin BDNF, we find that BDNF has antidepressant-like properties, as measured by the forced-swim test; furthermore, this is paralleled by the prevention of chronic stressinduced hippocampal atrophy in the transgenic mice. Paradoxically, the transgenic mice exhibit enhanced anxiety, which is correlated with increased spine-density on pyramidal cells in the basolateral amygdala, a finding that is consistent with results described in section a. This suggests that structural remodeling in the hippocampus and amygdala may serve as the substrate for depression and anxiety respectively. Thus, structural changes in the hippocampus and amygdala, caused by genetic manipulation of the same molecule BDNF, give rise to contrasting effects on anxiety and depressive symptoms, both of which are major behavioral correlates of stress disorders.
Collaborators: Susumu Tonegawa, Arvind Govindarajan, MIT; and
B. S. Shankaranarayana Rao, NIMHANS
b. Molecular mediators of spine plasticity.
Sharath Bennur, Amrita Kuthiala and Deepti Rao
Memory formation and consolidation are thought to involve changes in the number and structure of dendritic spines. In view of our findings on stress-induced spine formation, we are using transgenic/knockout mouse models to study molecular mechanisms that may mediate such structural plasticity. In one study, we observed that cortical neurons in transgenic mice, in which catalytic activity of p21-activated kinase (PAK) is impaired in the postnatal forebrain, have fewer dendritic spines compared to wild-type controls. By contrast, hippocampal spine morphology was normal. Strikingly, these mice were deficient in longterm consolidation, but not acquisition, of hippocampal memory. These results provide evidence for a pivotal relationship between cortical spine morphology and consolidation of long-term memory. These mice are now being used to further investigate the links between anxiety and stress-induced spine formation in the BLA. We are also investigating the role of the serine protease tissue-plasminogen activator (tPA) in stress-induced spine plasticity and anxiety-like behavior. We find that mice in which the tPA gene has been disrupted, the effects of chronic stress on spine-density are significantly attenuated in the medial amygdala.
Collaborators: Bruce McEwen and Robert Pawlak, Rockefeller University. Susumu Tonegawa and Mansuo Hayashi, MIT, USA and B. S. Shankaranarayana Rao, NIMHANS, Bangalore
c. Fragile X mental retardation and metabotropic glutamate receptor dependent plasticity.
Sharath Bennur, Deepti Rao, Amrita Kuthiala and Aparna Suvrathan
Fragile X syndrome, a common form of mental retardation, is caused by the absence of the FMR1 gene. Previous studies have demonstrated the Fmr1- nockout (KO) mouse to be an excellent animal model for investigating many of the symptoms observed in human fragile X syndrome. A growing body of evidence, from studies using the Fmr1-KO mice as well as rats, has contributed to the “mGluR theory” which proposes that various aspects of fragile X syndrome are a consequence of exaggerated metabotropic glutamate receptor (mGluR) function. A majority of the findings that have contributed to the mGluR theory are derived from studies focused primarily on the hippocampus. However, some of the key symptoms of fragile X syndrome include abnormal affective behavior, such as anxiety. Although most of these emotional or mood-related symptoms are likely to involve changes in amygdalar function, there are no comprehensive studies that have examined amygdalar plasticity in the context of the mGluR theory. Therefore, we are investigating the synaptic basis of affective symptoms in fragile X syndrome using both rats and Fmr1-KO mice. To this end we have discovered a novel form of LTP that is induced by chemical activation of mGluRs in the LA, which is in contrast to mGluR-dependent long-term synaptic depression (LTD) previously observed in the hippocampus.
Collaborators: Mark Bear, Gordon Smith and Gul Dolen, MIT, USA