
Dr. Shruthi Viswanath - Research
Integrative structure determination
Structures of several large protein complexes and assemblies are difficult to obtain using traditional experimental methods. Integrative structure determination fills this gap; various types of experimental data are combined along with principles from physics, statistical inference, and prior models to obtain the structure. The different sources of input information may span multiple scales (for example, X-ray data is at the atomic scale, while FRET distances are at the domain scale). However, these various sources can provide complementary information (for example, EM maps may provide the shape of a complex while chemical crosslinks may provide the orientation of binding interfaces). We rigorously incorporate each type of input information while accounting for its uncertainty in a Bayesian inference framework. This allows us to incorporate data that is sparse, noisy, ambiguous, and from heterogenous samples.
Integrative structures of chromatin-modifying assemblies
We recently determined the structures of sub-complexes of the Nucleosome Remodeling and Deacetylase (NuRD) complex, a chromatin-modifying assembly that regulates gene expression and DNA damage repair. It is conserved across plant and animal species and expressed in most metazoan tissues. However, its structure is hard to characterize experimentally. Using Bayesian integrative structure determination, we combined information from published SEC-MALLS, DIA-MS, XLMS, negative stain EM, X-ray crystallography, and NMR spectroscopy, secondary structure and homology predictions. The integrative structures were corroborated by independent cryo-EM maps, biochemical assays, and known cancer-associated mutations.
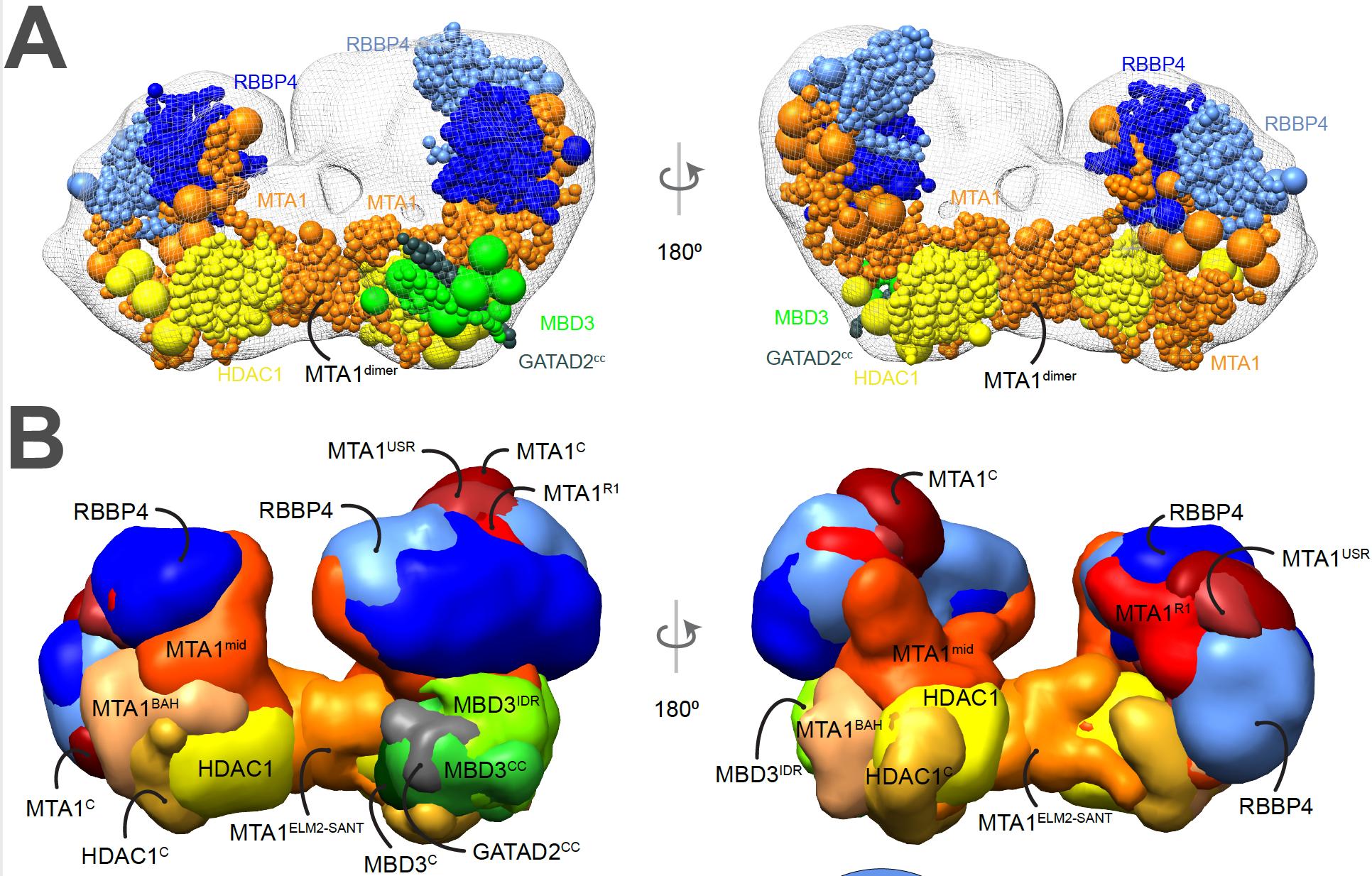
Integrative structure of the nucleosome deacetylase (NuDe) complex.
We are applying similar methods to study assemblies at cell-cell junctions, cytoskeletal, and centriolar assemblies.
Improving integrative modeling methods
We developed a method to optimize the coarse-grained representation for integrative models in IMP (https://integrativemodeling.org). The method, NestOR, uses Bayesian model selection to select from multiple user-specified candidate representations.

Coarse-grained representations are scored by their model evidence and sampling efficiency.
PrISM is our recently developed method to identify high and low precision regions in an ensemble of integrative models of large macromolecular assemblies. It is now used in the pipeline for validating integrative models in the PDB (worldwide Protein Data Bank).
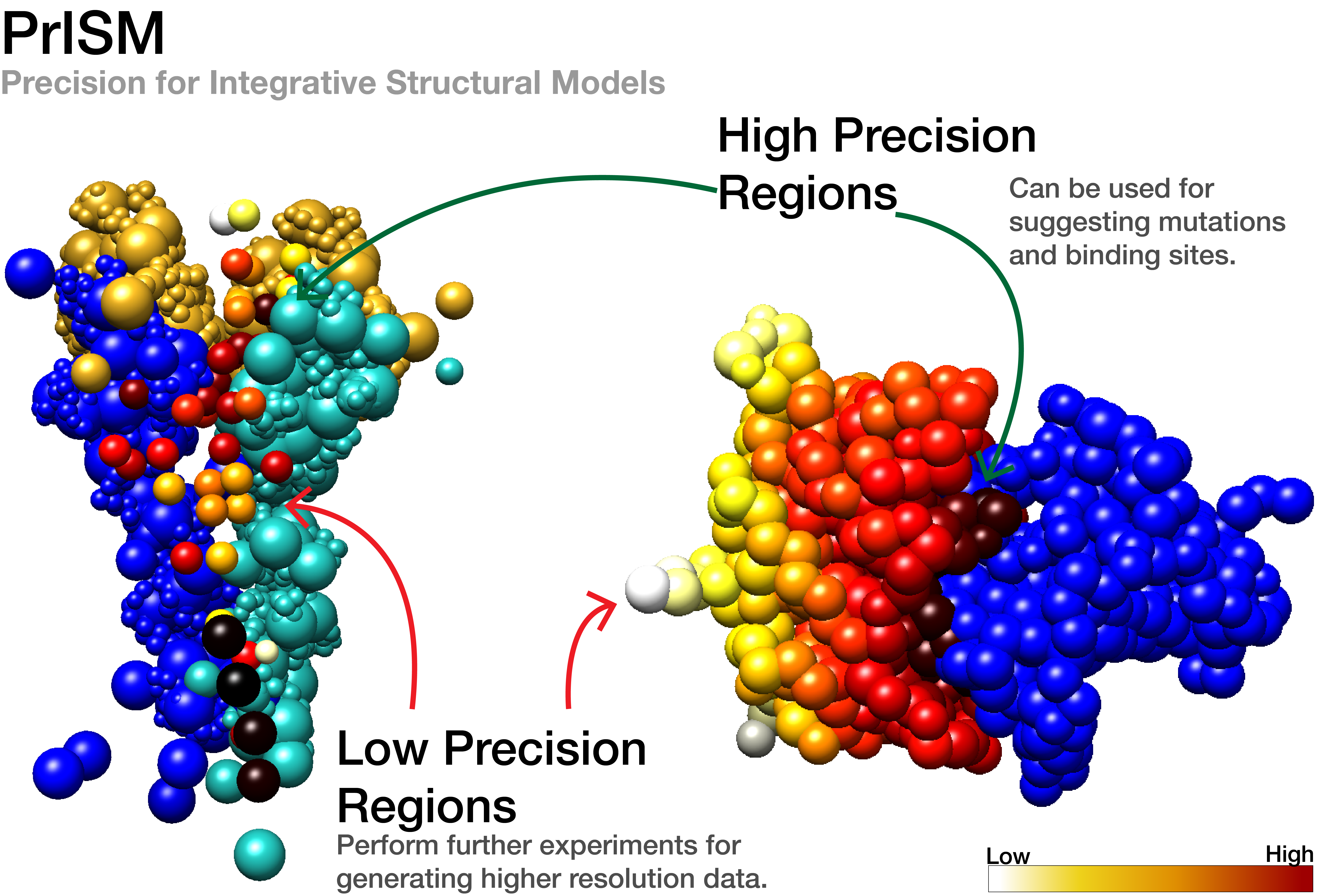
Annotating precision for integrative models