
Dr. Mrinalini Puranik - Research
Research Report 2005-2007:
Structure and dynamics of nucleic acid binding proteins
Nucleic acid-protein interactions are central to cellular function. Our lab is studying these interactions in two important cellular contexts: the maintenance of genomic integrity via DNA repair and the production of mononucleotides. Both processes involve highly specific recognition of the nucleic acid substrate and subsequent catalysis by the enzyme.
Anti-cancer drugs, normal cellular metabolism, and environmental toxins cause chemical modifications in DNA and RNA. These changes are reversed by cellular DNA repair machinery that identifies the presence of DNA damage and eliminates it by one of several ways. The most common repair strategy for single base modifications is base excision repair (BER) in which the damaged base is identified and removed by enzymes known as DNA repair glycosylases. Modifications due to oxidation and some types of alkylation of nucleobases are repaired by this method. Unique to the repair of alkylation damage is in-situ repair in which the base is restored to its original form without excision from the DNA. The alkylation of guanine by the anti-cancer drug temozolomide to form O6-methylguanine is countered by the human enzyme, MGMT by this process and is a cause of resistance to chemotherapy. MGMT transfers a methyl group from the nucleobase to a cysteine residue and is subsequently targeted for degradation. In an alternative form of in-situ repair, enzymes catalyse removal of the alkyl group and release the corresponding aldehyde e.g., theE.coli protein AlkB and the human proteins ABH2 and ABH3 which repair methylated adenine and cytosine.
Another important cellular process is the recycling of free, undamaged nucleobases into nucleotides carried out by enzymes called phosphoribosyl transferases (PRTases). Although DNA nucleobase repair and nucleotide salvage are distinct functions, they present similar challenges to the enzymes involved. Each nucleobase substrate must be identified with high specificity while still maintaining the ability to catalyze a wide range of substrates. From the available crystal structures it appears to be achieved by having a protein architecture that permits conformational flexibility rather than a rigid, preassembled active-site (See Figure). The common steps in the two types of nucleobase-protein interaction are then: the recognition of the substrate, followed by the assembly of an appropriate active-site for catalysis. Our research aims to understand the mechanistic details of this process.
Instrumentation Development: Fundamental to understanding these mechanisms is the ability to study the structure of proteins and nucleic acids in solution. We have developed ultraviolet resonance Raman spectroscopy (UVRR) of nucleobases as a probe of the protein active-site. Our experiments have demonstrated for the first time that the shifts in the observed Raman spectrum of the nucleic acid-protein complex are an excellent probe of their structure. We are now exploring deeper into the ultraviolet region to directly probe the proteins and obtain complementary information.
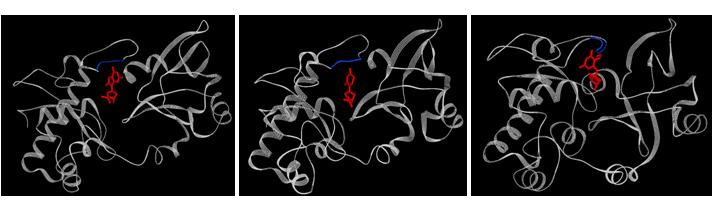
Figure: Bacterial formamidopyrimidine glycosylase (FPG) bound to various substrates. Left: Bacillus stearothermophilus FPG bound to 8-oxoguanine (PDB ID: 1R2Y); middle: Bacillus stearothermophilus FPG bound to 5,6-dihydrouracil (PDB ID: 1R2Z); right: Lactococcus lactis FPG bound to 2,6-diamino-4-hydroxy-5-formamidopyrimidine (PDB ID: 1XC8). A few residues of the protein have been deleted to give a better view of the nucleotide. Residues 4-8 (left), residues 4-12 (middle), residues 3-8 (right) of the protein are not shown to provide a clear view of the nucleotide.
1. Molecular mechanism of multiple substrate specificty of the base excision repair enzyme formamidopyrimidine glycosylase (fpg)
Oxidation of DNA by reactive oxygen species produced during the normal metabolism of the cell leads to covalent modification of nucleobases. These modified nucleobases are mutagenic in nature and their presence is implicated in cancer, neurodegenerative disorders and aging. A well-known, major product of DNA oxidation in cells is 8-oxoguanine. The deleterious effects of 8-oxoguanine are combated by formamidopyrimidine glycosylase (FPG), one of the key enzymes in the base excision repair pathway (Figure 1). FPG is highly specific in distinguishing 8-oxoguanine from its normal counterpart, guanine, which differs by only two atoms. In addition to 8-oxoguanine, FPG also recognizes and excises other modifications of guanine, uracil and cytosine. This promiscuity is characteristic of glycosylases and is hypothesized to originate in the conformational flexibility of their active-sites. Indeed, the crystal structures of FPG from different organisms in free and substrate bound forms (opposite page) reveal considerable conformational changes in the protein structure upon the binding of each type of substrate.
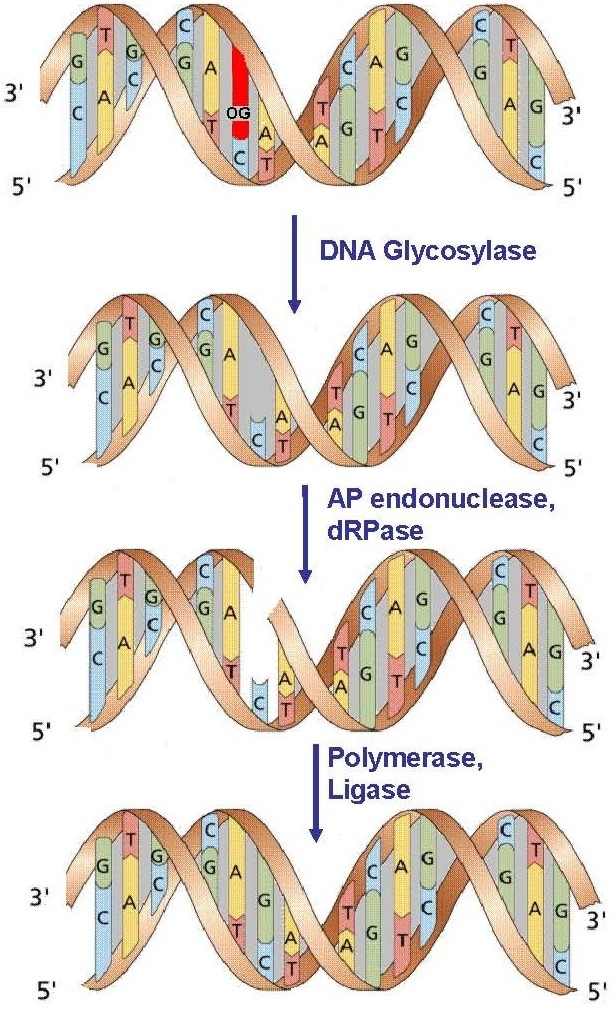
Figure 1. The Base Excision Repair pathway.
Elucidation of the precise mechanism of the recognition of a damaged nucleobase, formation of the active-site and subsequent catalysis requires an understanding of the nucleobase-protein interaction in solution. We are studying the nature of FPG-DNA interaction using UVRR. As a first step, we have studied the structure of free 8-oxoguanine in solution using UVRR. Raman spectra obtained with laser excitation at 260 nm of guanine and 8-oxoguanine show remarkably different relative intensities indicative of the difference in the structures of the electronic excited states responsible for the absorption at 260 nm in the two molecules. We have shown that UVRR can be used as a base-specific probe to distinguish between guanine and 8-oxoguanine (Figure 2). The contentious structure of 8-oxoguanine is unequivocally established to be that of the diketo tautomer in solution from our experiments, quantum chemical calculations and normal mode analysis. Having established the specific Raman signatures of the oxidized base, we are now using the nucleobase as a probe of the FPG active-site. Experiments involve probing the complex of FPG with 8-oxoguanine containing DNA.
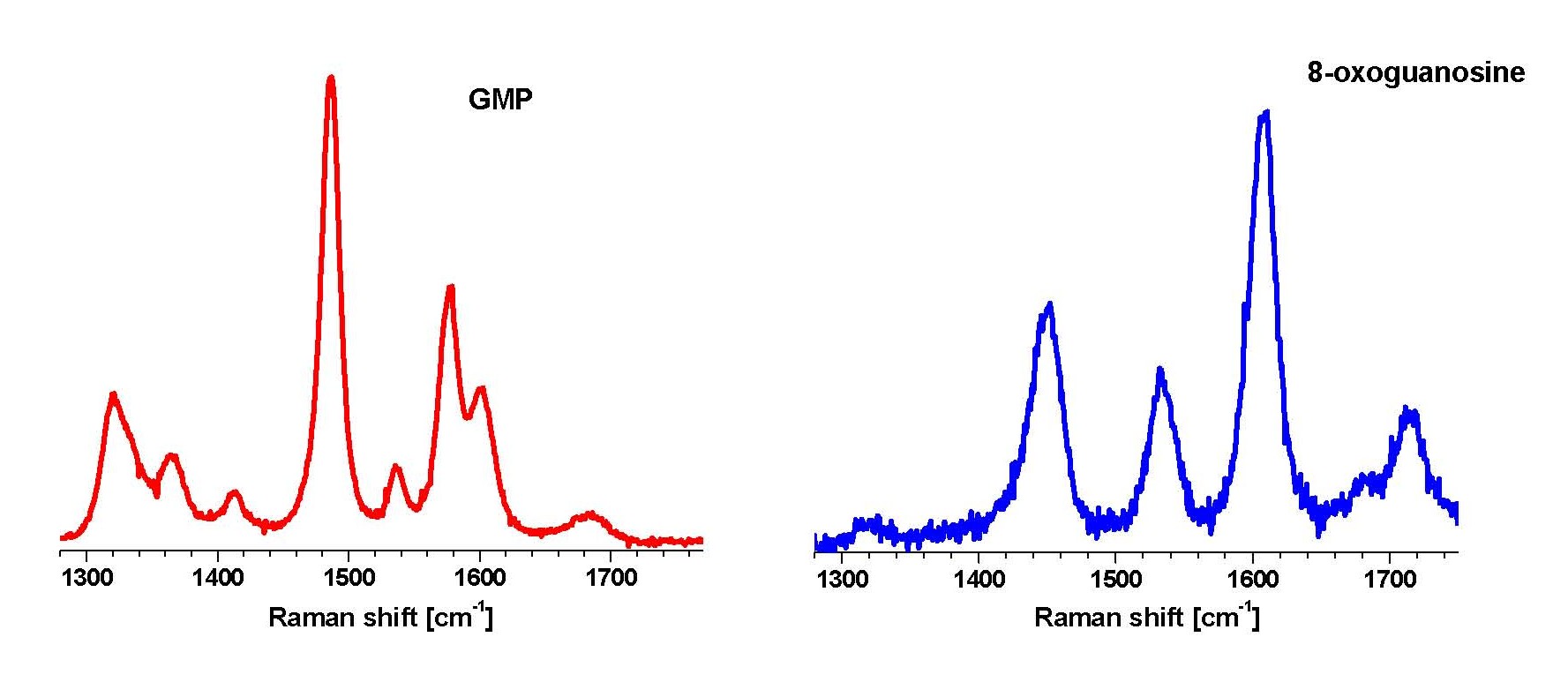
Figure 2. Ultra-violet resonance Raman spectra of GMP and its oxidized form, 8-oxoguanosine, in water, pH 7.0, obtained with 260 nm excitation wavelength.
2. On-site repair of alkylated DNA by E.coli AlkB and its human analogues
Alkylating chemicals are widely used as chemotherapeutic drugs in the treatment of cancer, e.g., carmustine, temozolomide, etc. S-adenosylmethionine present in cells is also a potential methylating agent. These and other alkylating agents in the environment damage DNA by adding alkyl groups to nucleobases. Alkylation of nucleobases alters their hydrogen bonding properties leading to cytotoxicity and mutagenicity. Similar modifications can be expected to occur in RNA nucleobases as well.
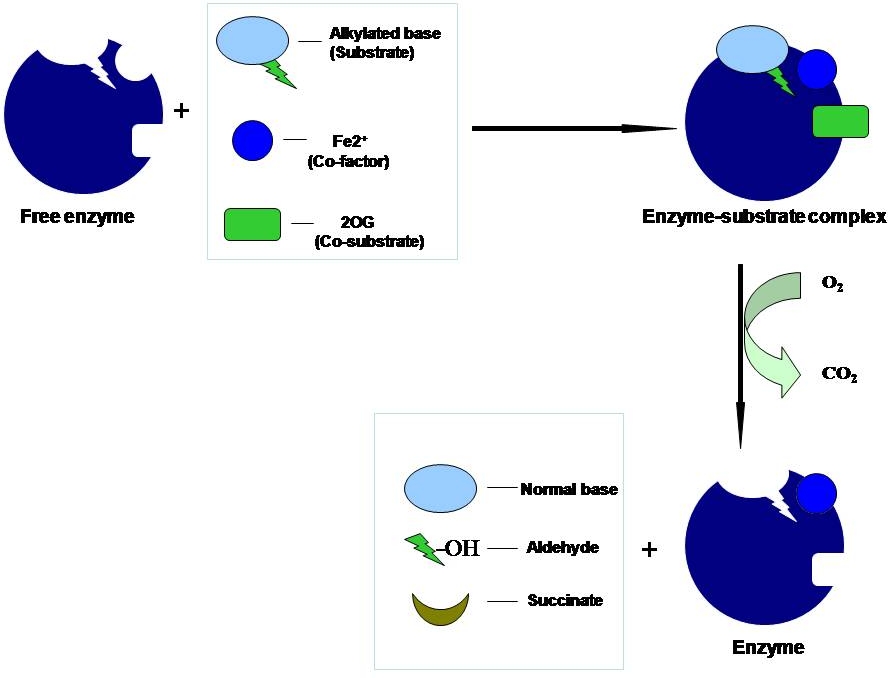
E. coli AlkB (Figure 3) and its human homologues ABH2 and ABH3 bring about direct damage reversal of alkylated single and double stranded DNA and RNA in a reaction that requires Fe(II) as a cofactor, α-ketoglutarate as co-substrate and oxygen (Figure 4). AlkB enzyme repairs alkylation lesions such as N1-methyladenine (1meA) and N3-methylcytosine efficiently and N1-methylguanine and N3-methylthymine with lower efficiency. Apart from these small lesions, AlkB removes larger alkyl groups such as ethyl and propyl groups and exocyclic ethano adducts like 1,N6-ethanoadenine and 3,N4-ethanocytosine. These lesions are formed only in single stranded DNA during replication and transcription as these sites are involved in base pairin10g between complementary strands in a double stranded DNA. How does AlkB accommodate and catalyze both small and large lesions?
The crystal structure of substrate-bound AlkB solved recently by Hunt and coworkers (Nature 2006) provided detailed insight into the mechanism of AlkB action. The Fe(II)-oxoglutarate dioxygenase core matched other superfamily members but the trinucleotide alkylated substrate showed a unique fold designed to hold the 1meA in correct orientation. A conformationally flexible ‘lid’ was found that completes the assembly of the active-site pocket when the substrate binds to AlkB. The transport of molecular oxygen, essential for the reaction appears to be through a tunnel connected to the active site. The crystals were prepared and examined in the absence of molecular oxygen to prevent the catalytic reaction. However, when oxygen was provided, the reaction was found to be inefficient in the crystal. This suggests the possibility that the structure in solution is different and that protein dynamics may modulate the chemistry. These crystal structures have thrown open several interesting questions about the action of AlkB, the role of protein dynamics and the assembly of the active site pocket for different substrates. We will now address these for the first time using resonance Raman spectroscopic approaches developed in our laboratory.
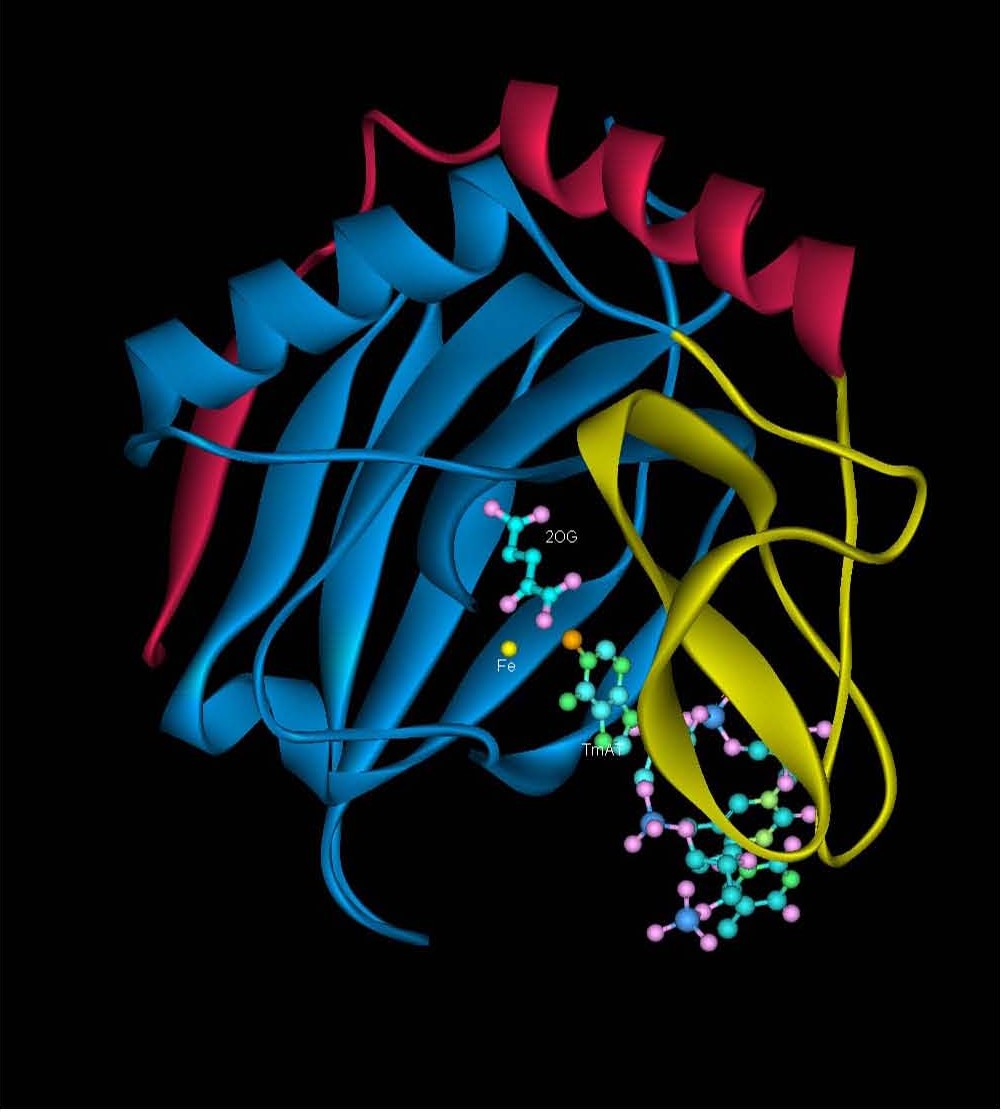 |
Figure 3: Crystal structure of AlkB (PDB ID: 2FD8) in anaerobic conditions showing the Fe(II), nucleobase and 2-oxoglutarate (2-OG) and methylated trinucleotide d(T-meA-T) bound to it. |
| Figure 4: Mechanism of alkylation damage reversal by AlkB enzyme in the presence of the Fe(II) co-factor and the co-substrate (2-oxoglutarate) with the consumption of one molecule of oxygen and release of carbon-di-oxide and corresponding aldehyde. |
 |
3. Understanding the different substrate specificities of human and Plasmodium falciparum Hypoxanthine guanine phosphoribosyl transferase (HGPRT)
Nucleotide synthesis in humans is carried out by the de novo synthesis pathway, where nucleotides are synthesized from small molecules present in the cell, and the salvage pathway which involves recycling of previously metabolized intermediates, e.g. purines. Some protozoan parasites likePlasmodium falciparum, the malarial parasite, possess only the salvage pathway for nucleotide synthesis. Inhibitors for this pathway would block nucleotide synthesis and are, thus, a target for chemotherapy against diseases caused by these parasites. The enzyme HGPRT that is a part of the purine salvage pathway is one such therapeutic target. Our current interest is to elucidate the catalytic mechanism of this enzyme and to understand differences between the human andPlasmodium enzymes.
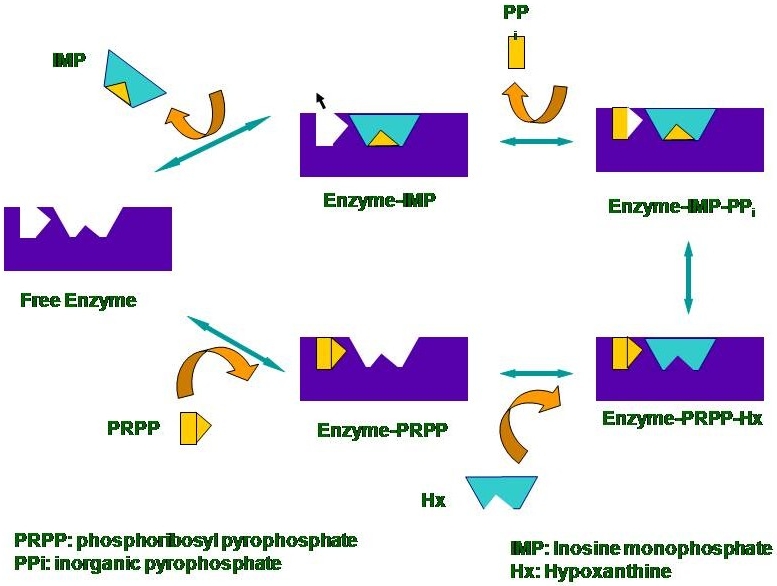
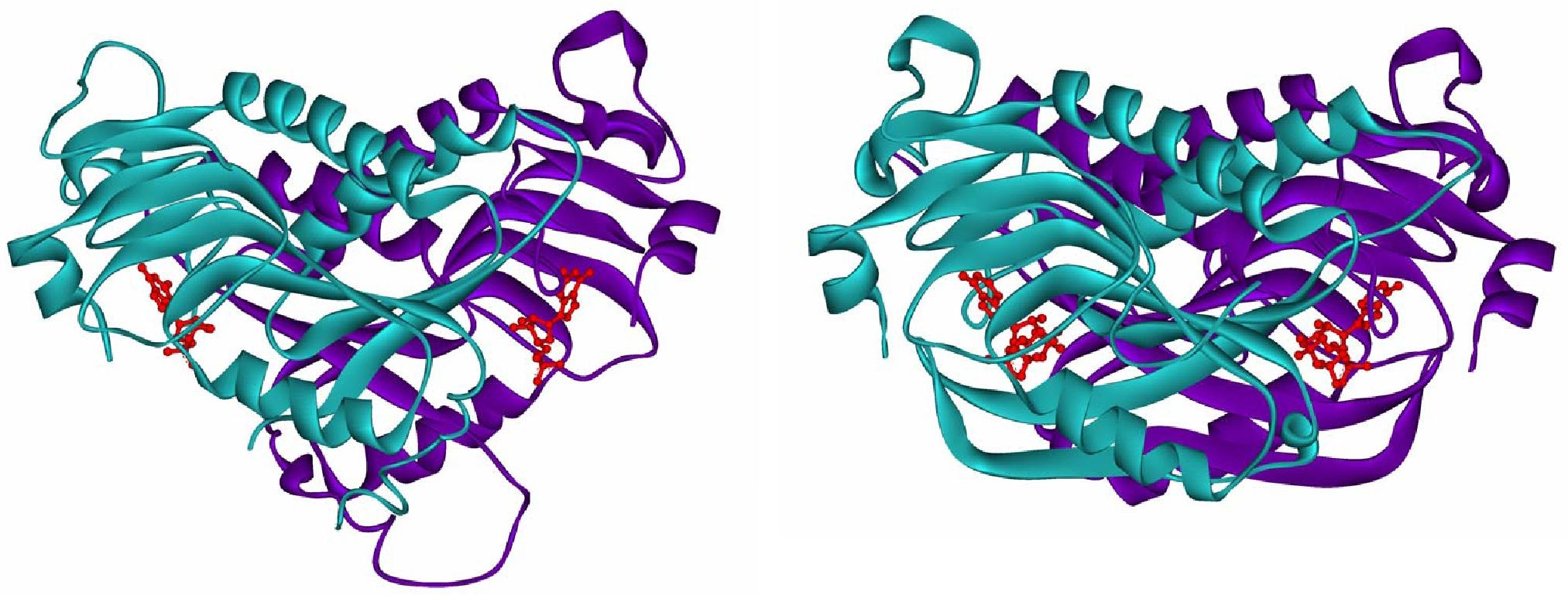
For these studies, we have developed a first application of UVRR spectroscopy to observe the nucleotide specifically while it is bound to the enzyme. The human and malarial enzymes exhibit differing substrate specificities with respect to xanthine. The human as well as Plasmodium falciparum enzymes catalyze the conversion of hypoxanthine and guanine into their nucleotide counterparts inosine monophosphate and guanosine monophosphate, respectively (Figure 5). However, the malarial enzyme catalyses the conversion of xanthine to xanthosine monophosphate as well while the human enzyme does not. The crystal structures currently available do not explain this difference between the two enzymes (Figure 6). With an aim to understand the origin of this substrate specificity, we have examined the end-product complexes of huHGPRT and pfHGPRT spectroscopically. Our current experiments of the enzyme-end-product complexes demonstrate that the environment of the nucleobase in the two enzymes has subtle differences. Raman spectra of the nucleotides bound to the enzymes show shifts that are found to correlate with the published crystal structures of the enzymes. We plan to carry out experiments with various available site mutants of the enzyme to elucidate the origin of substrate specificity and mechanism of catalysis.
| Figure 5: The proposed catalytic mechanism of the human enzyme. The last rate limiting step of the product binding to the enzyme by incubating the enzyme with a large excess of the product was studied with UVRR. |
 |
| Figure 6: Crystal structures of human HGPRT with bound GMP, (PDB ID: IHMP) left; and P. falciparum HGPRT bound to the transition state inhibitor immucillin, right. (PDB ID: 1CJB) |
 |
While the Raman shifts observed experimentally will provide a qualitative measure of the protein-nucleotide interaction, we are using a computational approach to obtain a quantitative interpretation of the observed shifts. Currently, we are carrying out ab initio and density functional theoretical calculations using co-ordinates from the available crystal structure of the enzyme to simulate the protein-nucleotide interaction. The computed structure of the free base (from HF/DFT) will be compared with the structure of the base in the active-site of the protein. The changes in structure due to interaction with the amino acids of the protein will be correlated with observed changes in the corresponding Raman spectra. We will further extend these studies to model other substrates and end-products for which crystal structures are not yet available.
Collaborator: Hemalatha Balaram, Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore.